compute stress/atom command¶
Syntax¶
compute ID group-ID stress/atom general_keyword general_values keyword ...
Examples¶
compute 1 mobile stress/atom
compute 1 all stress/atom pair bond
Description¶
Define a computation that computes the symmetric per-atom stress tensor for each atom in a group. The tensor for each atom has 6 components and is stored as a 6-element vector in the following order: xx, yy, zz, xy, xz, yz. See the compute pressure command if you want the stress tensor (pressure) of the entire system.
The stress tensor for atom I is given by the following formula, where a and b take on values x,y,z to generate the 6 components of the symmetric tensor:
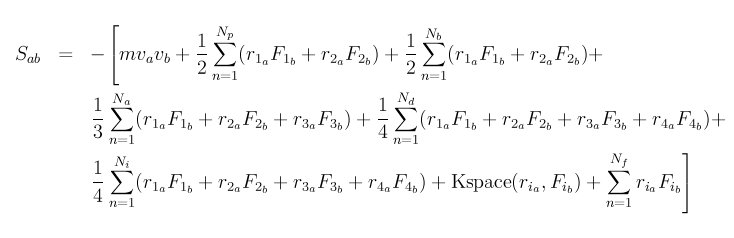
The first term is a kinetic energy contribution for atom I. The
second term is a pairwise energy contribution where n loops over the
Np neighbors of atom I, r1 and r2 are the positions of the 2
atoms in the pairwise interaction, and F1 and F2 are the forces on
the 2 atoms resulting from the pairwise interaction. The third term
is a bond contribution of similar form for the Nb bonds which atom
I is part of. There are similar terms for the Na angle, Nd
dihedral, and Ni improper interactions atom I is part of. There
is also a term for the KSpace contribution from long-range Coulombic
interactions, if defined. Finally, there is a term for the Nf
fixes that apply internal constraint forces to atom I.
Currently, only the fix shake and fix rigid commands contribute to this term.
Warning
For granular systems, this formular neglects the contribution of average velocity in the kinetic energy contribution. This is corrected in the compute ave/euler command (currently no doc available).
As the coefficients in the formula imply, a virial contribution
produced by a small set of atoms (e.g. 4 atoms in a dihedral or 3
atoms in a Tersoff 3-body interaction) is assigned in equal portions
to each atom in the set. E.g. 1/4 of the dihedral virial to each of
the 4 atoms, or 1/3 of the fix virial due to SHAKE constraints applied
to atoms in a a water molecule via the fix shake
command.
If no extra keywords are listed, all of the terms in this formula are included in the per-atom stress tensor. If any extra keywords are listed, only those terms are summed to compute the tensor. The virial keyword means include all terms except the kinetic energy ke.
Note that the stress for each atom is due to its interaction with all other atoms in the simulation, not just with other atoms in the group.
The dihedral_style charmm style calculates
pairwise interactions between 1-4 atoms. The virial contribution of
these terms is included in the pair virial, not the dihedral virial.
The KSpace contribution is calculated using the method in
(Heyes) for the Ewald method and by the methodology described
in (Sirk) for PPPM. The choice of KSpace solver is specified
by the kspace_style pppm command. Note that for
PPPM, the calcluation requires 6 extra FFTs each timestep that
per-atom stress is calculated. Thus it can significantly increase the
cost of the PPPM calculation if it is needed on a large fraction of
the simulation timesteps.
Note that as defined in the formula, per-atom stress is the negative of the per-atom pressure tensor. It is also really a stress*volume formulation, meaning the computed quantity is in units of pressure*volume. It would need to be divided by a per-atom volume to have units of stress (pressure), but an individual atom’s volume is not well defined or easy to compute in a deformed solid or a liquid. Thus, if the diagonal components of the per-atom stress tensor are summed for all atoms in the system and the sum is divided by dV, where d = dimension and V is the volume of the system, the result should be -P, where P is the total pressure of the system.
These lines in an input script for a 3d system should yield that result. I.e. the last 2 columns of thermo output will be the same:
compute peratom all stress/atom
compute p all reduce sum c_peratom[1] c_peratom[2] c_peratom[3]
variable press equal -(c_p[1]+c_p[2]+c_p[3])/(3*vol)
thermo_style custom step temp etotal press v_press
Output info¶
This compute calculates a per-atom array with 6 columns, which can be accessed by indices 1-6 by any command that uses per-atom values from a compute as input. See Section_howto 15 for an overview of LIGGGHTS(R)-PUBLIC output options.
The per-atom array values will be in pressure*volume units as discussed above.
Restrictions¶
none